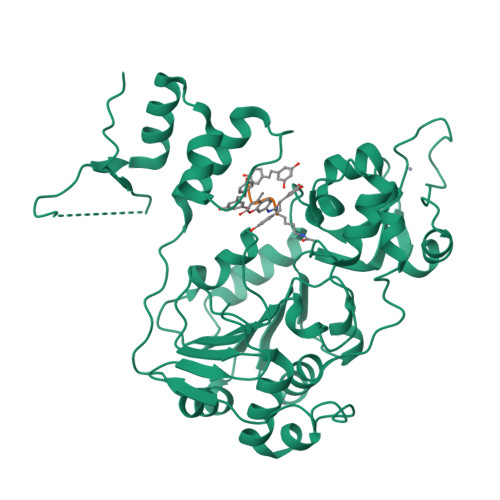
Docking
Docking
PDB: 1HSG
HIV-1 Protease with inhibitor
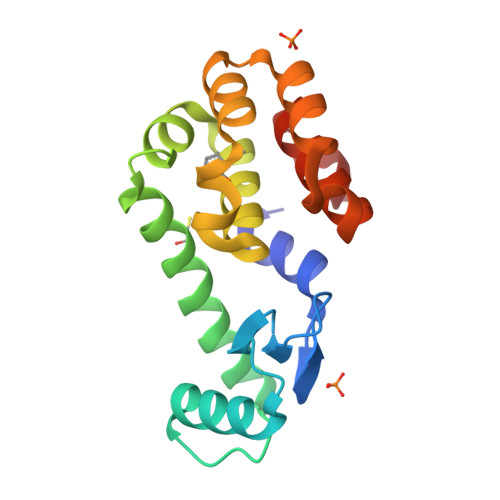
 NP target
NP target
PDB: 5BTR
SIRT1 — resveratrol complex
 Target
Target
PDB: 6LU7
SARS-CoV-2 Main protease
 Anti-inflammatory
Anti-inflammatory
PDB: 1PXX
COX-2 with ligand
Molecular docking
Where a molecule fits determines what it cures.
Bionics AI computes high-precision molecular docking at the binding pocket level. Given a protein structure and a natural compound SMILES, the platform identifies optimal binding poses, scores free energy of binding, and ranks candidates by estimated affinity — with full residue-level interaction maps.
Structures are energy-minimized via molecular dynamics before docking, ensuring pocket geometry reflects biologically realistic conditions, not idealized templates.
P2Rank
Binding pocket detection
Vina
AutoDock scoring engine
OpenMM
MD energy minimization
< 3 min
Fold → dock pipeline